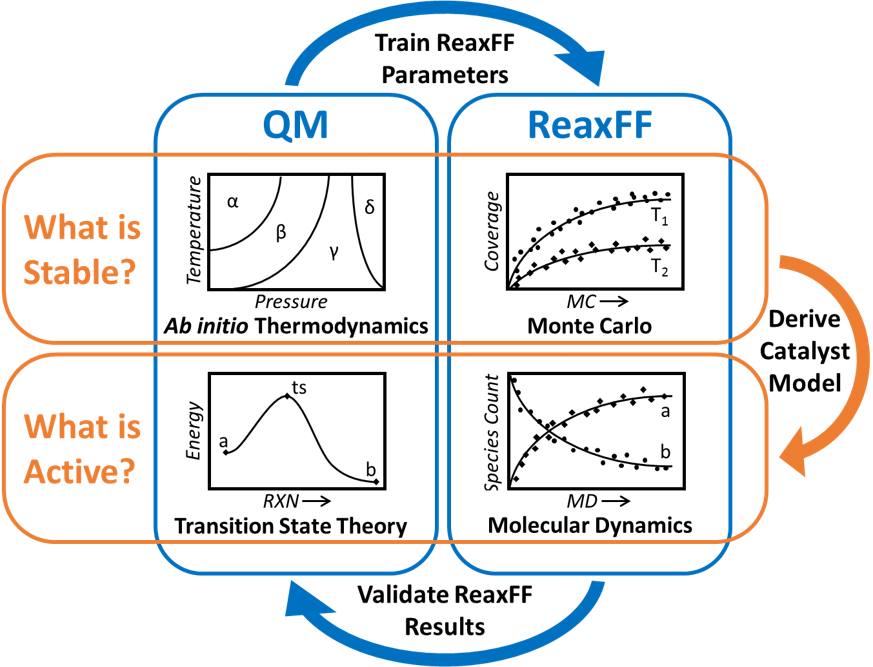
Multi-Physics Modeling: Catalysis and Materials for Clean Energy
The Senftle Group develops and applies computational modeling tools for assessing complex, multi-component catalysts at both the electronic and atomistic level. Particular focus is placed on developing fundamental structure-activity relationships informing the rational design of catalytic systems for efficient energy conversion, storage, and utilization.
Unraveling the Nature of Complex Metal-Support Interactions in Catalysis
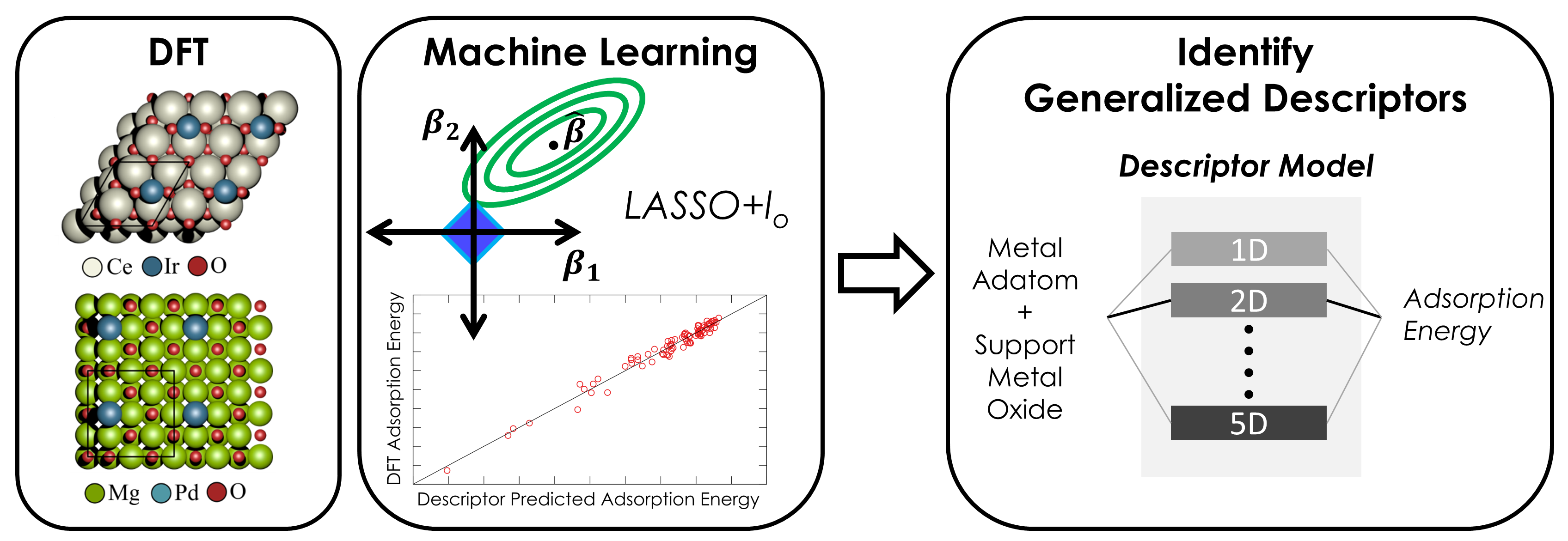
Heterogeneous catalysts featuring transition metal nanoparticles supported on oxide surfaces play an essential role in energy, environment, and chemical technologies. Synergistic interactions between the supported metal and the oxide surface can alter catalytic behavior and therefore must be understood at a fundamental level to tune overall catalytic activity, selectivity, and stability. Such interactions are complex, often resulting from the simultaneous action of multiple independent phenomena. We employ density functional theory (DFT) in concert with screening approaches derived from machine learning (ML) to identify generalized descriptors that control metal-support interactions. This approach provides predictive models that can identify useful modifications of the support or cluster morphology via the introduction of defects and dopants. Beyond catalysis, we develop methodologies for uncovering the fundamental properties responsible for governing charge transfer interactions in other multi-component systems, such as electrodes employed in photovoltaics, batteries, and fuel cells.
Catalysis for Environmental Applications
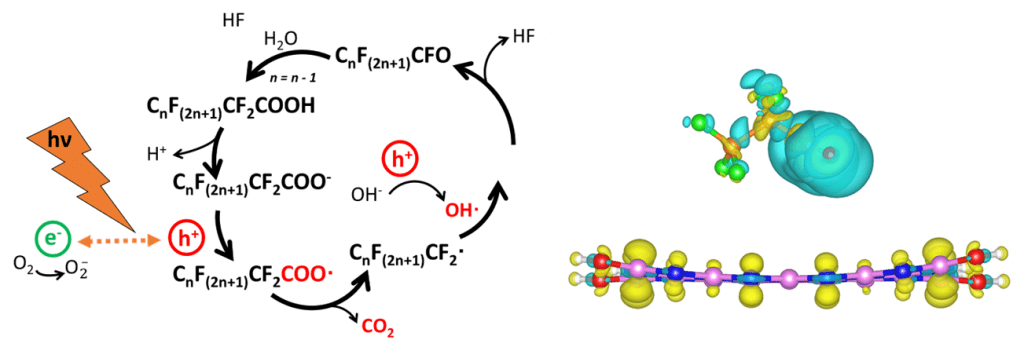
A key focus area of our group is the design of catalysts for water treatment and environmental applications. We are members of the NSF Nanosystems Engineering Research Center for Nanotechnology Enabled Water Treatment Systems (NEWT). In particular, we design catalysts for the treatment of per- and poly-fluorinated substances (PFAS, “forever chemicals”) and nitrate in drinking water. Specific project examples include (1) the use of simulation to determine photo-catalytic mechanisms for degrading PFAS and (2) the use of microkinetic modeling to determine reaction pathways during the nitrate reduction reaction.